Imaging near the quantum limit
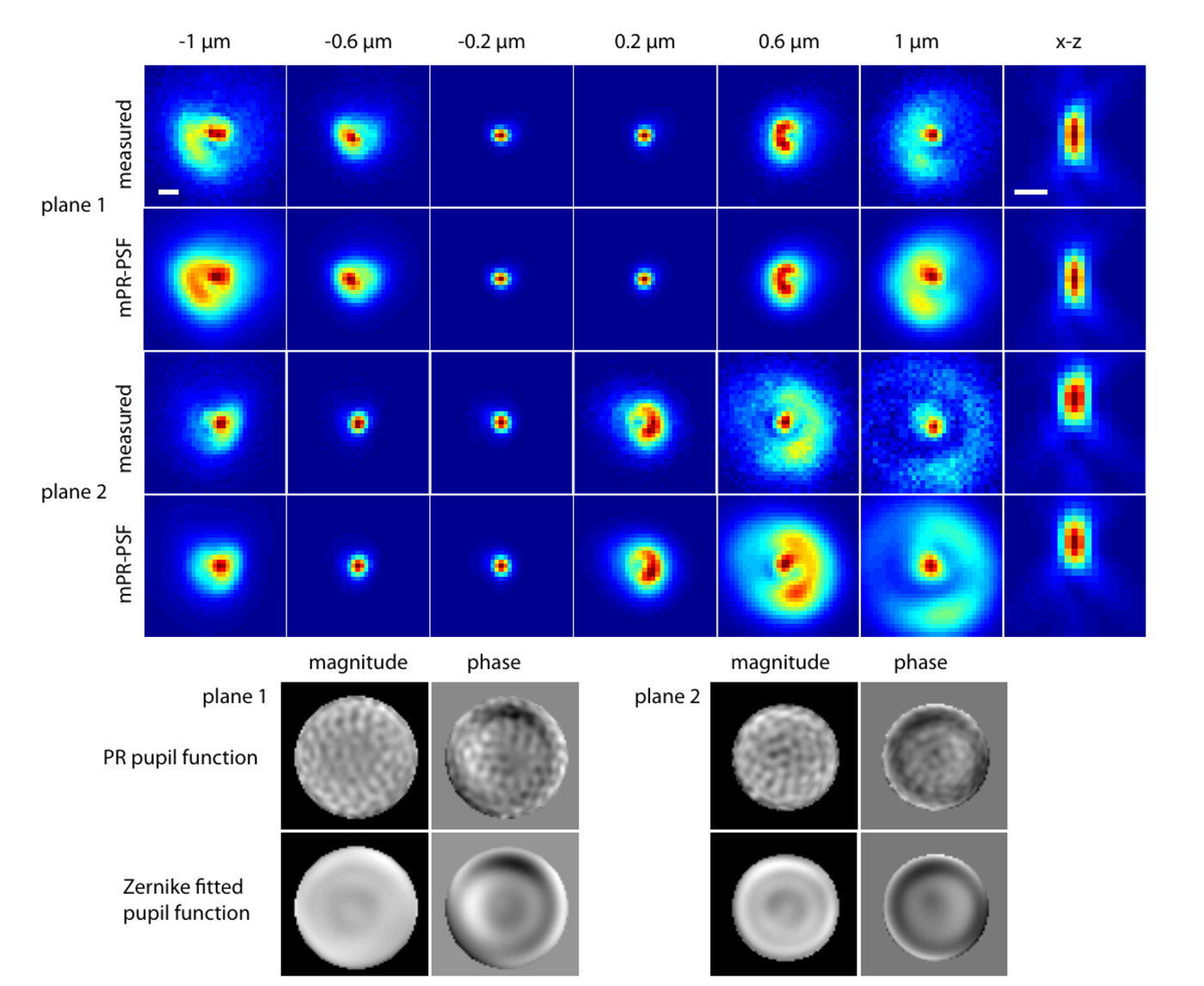
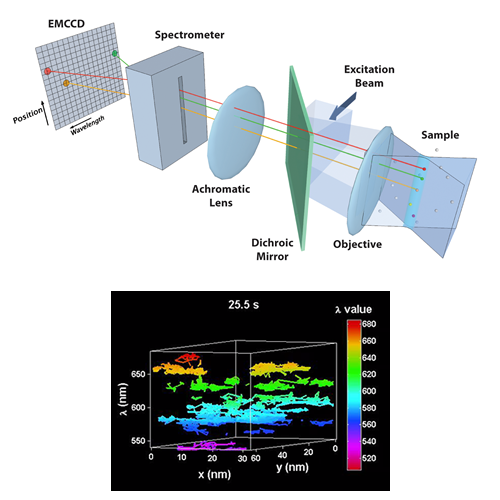
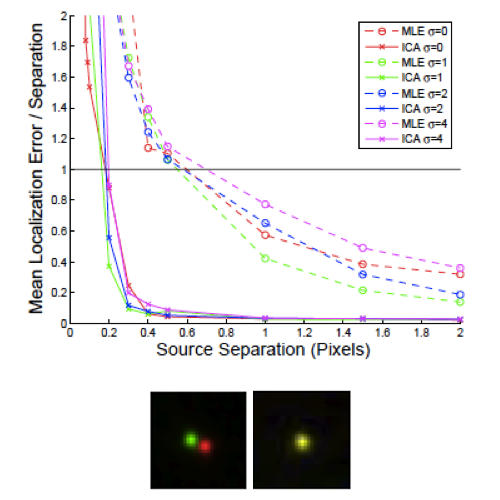
In 2016, quantum estimation theory upended Rayleigh's criterion: Tsang, Nair, and co-workers showed that “Rayleigh's curse,” the collapse of separation-estimation precision below the diffraction limit, is not fundamental, and that mode-sorting measurements such as SPADE and SLIVER can reach the quantum Cramér–Rao lower bound (QCRLB), the precision limit for any measurement quantum mechanics allows. But that theory, and its experimental demonstrations, assumed idealized scalar point sources, while the single fluorophores of super-resolution microscopy are freely rotating dipole emitters imaged through high-NA objectives, where the quantum limit was unknown. In Liu et al. (Physical Review A, 2026) we computed the QCRLB for two freely rotating dipoles using vectorial diffraction theory and found, contrary to scalar-model predictions, that standard SLIVER loses its advantage at high numerical aperture because dipole radiation mixes even and odd fields, degrading interference visibility. We introduced Polar-SLIVER, which uses a vortex wave plate to send the purely antisymmetric azimuthal polarization into the inversion interferometer, restoring non-divergent, near quantum-limited precision at any separation, and quantified the effects of background, detection bandwidth, and misalignment. Earlier, in Schodt et al. (Optics Express, 2023), we showed that an image inversion interferometer keeps its precision advantage over direct imaging across a wide range of aberrations and misalignments when pixelated detection is used.